15 RCircos zoomed heatmap, histogram, and points plot
Authors: Hongen Zhang Reviser: Tianze Cao
2025-06-05
Source:vignettes/15 Zoom_Plot_Demo.Rmd
15 Zoom_Plot_Demo.RmdRCircos Zoom Plot Demo
This demo plot human chromosome ideogram with zoomed heatmap plot, zoomed hitogram plot, and zoomed scatter(points) plot on outside of chromosome ideogram.
# Initialize the plot objects
library(RCircos);
data(UCSC.HG19.Human.CytoBandIdeogram);
cyto <- UCSC.HG19.Human.CytoBandIdeogram;
RCircos.Set.Core.Components(cyto.info=cyto, chr.exclude=NULL,
tracks.inside=10, tracks.outside=5);##
## RCircos.Core.Components initialized.
## Type ?RCircos.Reset.Plot.Parameters to see how to modify the core components.
RCircos.Set.Plot.Area();
RCircos.Chromosome.Ideogram.Plot();
# Zoom heatmap plot with defined track number
data(RCircos.Heatmap.Data);
min.value <- min(as.matrix(RCircos.Heatmap.Data[,5:10]));
max.value <- max(as.matrix(RCircos.Heatmap.Data[,5:10]));
RCircos.Heatmap.Plot(RCircos.Heatmap.Data, data.col=5,
side="in", track.num=1);
zoom.data <- RCircos.Get.Zoom.Data(RCircos.Heatmap.Data, name.col=4,
genomic.columns=3, target.gene="SP5", neighbor.genes=5);
zoom.range <- RCircos.Get.Zoom.Range(zoom.data, genomic.columns=3);
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range, total.genes=11,
area.length=0.25, fixed.width=TRUE, gene.width=NULL);
RCircos.Plot.Zoomed.Heatmap(zoom.data, data.col=5, zoom.pos=zoom.pos,
track.num=2, outside.pos=NULL, inside.pos=NULL,
min.value, max.value);
RCircos.Mark.Zoom.Area(zoom.range, track.num=2, zoom.pos=zoom.pos,
fill.color="red", inside.pos=NULL, outside.pos=NULL);
RCircos.Label.Zoom.Region(zoom.data, name.col=4, zoom.pos=zoom.pos,
text.size=0.6, track.num=3, outside.pos=NULL, inside.pos=NULL);
rm(zoom.data); rm(zoom.range); rm(zoom.pos);
rm(min.value); rm(max.value); rm(RCircos.Heatmap.Data);
# Zoom histogram plot with defined inside.pos and outside.pos
data(RCircos.Histogram.Data);
RCircos.Histogram.Plot(RCircos.Histogram.Data, data.col=4,
side="in", track.num=2);
data.rows <- which(RCircos.Histogram.Data$Chromosome=="chr4");
zoom.data <- RCircos.Histogram.Data[data.rows[1:11], ];
zoom.range <- RCircos.Get.Zoom.Range(zoom.data, genomic.columns=3);
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range, total.genes=11,
area.length=0.25, fixed.width=TRUE, gene.width=NULL);
track.pos <- RCircos.Get.Track.Positions("out", 2);
track.height <- track.pos[1] - track.pos[2];
in.pos <- track.pos[2];
out.pos <- in.pos + track.height*2;
RCircos.Plot.Zoomed.Histogram(zoom.data, data.col=4, zoom.pos=zoom.pos,
track.num=NULL, inside.pos=in.pos, outside.pos=out.pos);
RCircos.Mark.Zoom.Area(zoom.range, track.num=NULL, zoom.pos=zoom.pos,
fill.color="green", outside.pos=out.pos, inside.pos=in.pos);
rm(zoom.data); rm(zoom.range); rm(zoom.pos);
rm(data.rows); rm(track.pos); rm(track.height);
rm(in.pos); rm(out.pos); rm(RCircos.Histogram.Data);
# Zoom point(scatter) plot with defined inside.pos and outside.pos
data(RCircos.Scatter.Data);
RCircos.Scatter.Plot(RCircos.Scatter.Data, data.col=5, track.num=3,
side="in", by.fold=1);
data.col <- 5;
min.value <- min(as.matrix(RCircos.Scatter.Data[, data.col]));
max.value <- max(as.matrix(RCircos.Scatter.Data[, data.col]));
data.rows <- which(RCircos.Scatter.Data$Chromosome=="chr5");
zoom.data <- RCircos.Scatter.Data[1160:1171, ];
zoom.data["PlotColors"] <- rainbow(nrow(zoom.data));
zoom.range <- RCircos.Get.Zoom.Range(zoom.data, genomic.columns=3);
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range, total.genes=11,
area.length=0.25, fixed.width=TRUE, gene.width=NULL);
# Manually move zoome area a little forward to avoid overlap
zoom.pos <- zoom.pos + 1500;
track.pos <- RCircos.Get.Track.Positions(side="out", track.num=3);
track.height <- track.pos[1] - track.pos[2];
in.pos <- track.pos[2];
out.pos <- in.pos + track.height * 2;
RCircos.Plot.Zoomed.Scatters(zoom.data, data.col=5,
track.num=NULL, zoom.pos=zoom.pos,
min.value=min.value, max.value=max.value,
point.type=19, with.size=TRUE, point.scale=1,
inside.pos=in.pos, outside.pos=out.pos);
RCircos.Mark.Zoom.Area(zoom.range, track.num=NULL, zoom.pos=zoom.pos,
fill.color="yellow", inside.pos=in.pos, outside.pos=out.pos);
rm(zoom.data); rm(zoom.range); rm(zoom.pos);
rm(data.col); rm(min.value); rm(max.value);
rm(data.rows); rm(track.pos); rm(track.height);
rm(in.pos); rm(out.pos); rm(RCircos.Scatter.Data);
# Zoomed-in gene connectors and gene labels with defined track number
data(RCircos.Heatmap.Data);
data.rows <- which(RCircos.Heatmap.Data$Chromosome=="chr7");
zoom.data <- RCircos.Heatmap.Data[5580:5590, 1:4];
zoom.range <- RCircos.Get.Zoom.Range(zoom.data, genomic.columns=3);
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range, total.genes=11,
area.length=0.25, fixed.width=TRUE, gene.width=NULL);
RCircos.Plot.Zoomed.Gene.Connectors(zoom.data, zoom.pos=zoom.pos,
track.num=1, line.width=1, outside.pos=NULL, inside.pos=NULL);
# Zoomed gene connector will use three tracks so next track number
# must be three more
RCircos.Label.Zoom.Region(zoom.data, name.col=4, zoom.pos=zoom.pos,
text.size=0.7, track.num=4, outside.pos=NULL, inside.pos=NULL);
rm(zoom.data); rm(zoom.range); rm(zoom.pos); rm(data.rows);
rm(RCircos.Heatmap.Data);
# Zoomed-in parallel line plot with defined in and out plot positions
data(RCircos.Tile.Data);
RCircos.Tile.Plot(RCircos.Tile.Data, track.num=4, side="in");## Tiles plot may use more than one track. Please select correct area for next track if necessary.
data.rows <- which(RCircos.Tile.Data$Chromosome=="chr18");
zoom.data <- RCircos.Tile.Data[data.rows, ];
zoom.range <- RCircos.Get.Zoom.Range(zoom.data, genomic.columns=3)
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range,
total.genes=5, area.length=0.06, gene.width=NULL);
track.pos <- RCircos.Get.Track.Positions(side="out", track.num=3);
track.height <- track.pos[1] - track.pos[2];
in.pos <- track.pos[2];
out.pos <- in.pos + track.height * 2;
RCircos.Plot.Zoomed.Parallel.Lines(zoom.data, zoom.pos=zoom.pos,
track.num=NULL, genomic.cols=3, line.width=3,
inside.pos=in.pos, outside.pos=out.pos, outline=TRUE);
RCircos.Mark.Zoom.Area(zoom.range, track.num=NULL, zoom.pos=zoom.pos,
fill.color="yellow", inside.pos=in.pos, outside.pos=out.pos)
rm(zoom.data); rm(zoom.range); rm(zoom.pos); rm(data.rows);
rm(track.pos); rm(track.height); rm(in.pos); rm(out.pos);
rm(RCircos.Tile.Data);
# Zoomed-in tile plot with defined inside and outside plot positions
data(RCircos.Tile.Data);
data.rows <- which(RCircos.Tile.Data$Chromosome=="chrX");
zoom.data <- RCircos.Tile.Data[data.rows, ];
zoom.range <- RCircos.Get.Zoom.Range(zoom.data, genomic.columns=3);
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range,
total.genes=length(data.rows), area.length=0.08,
fixed.width=FALSE, gene.width=NULL);
track.pos <- RCircos.Get.Track.Positions(side="out", track.num=3);
track.height <- track.pos[1] - track.pos[2];
in.pos <- track.pos[2];
out.pos <- in.pos + track.height * 3;
layers <- RCircos.Get.Plot.Layers(RCircos.Tile.Data, genomic.columns=3);
RCircos.Plot.Zoomed.Tiles(zoom.data, zoom.pos=zoom.pos,
genomic.cols=3, layers=max(layers), track.num=NULL,
inside.pos=in.pos, outside.pos=out.pos, border.col=NULL);
RCircos.Mark.Zoom.Area(zoom.range, track.num=NULL, zoom.pos=zoom.pos,
fill.color="green", inside.pos=in.pos, outside.pos=out.pos);
rm(zoom.data); rm(zoom.range); rm(zoom.pos); rm(data.rows);
rm(track.pos); rm(track.height); rm(in.pos); rm(out.pos);
rm(RCircos.Tile.Data);
# Zoomed-in chromosome ideogram tick plot with defined
# inside and outside plot positions
zoom.range <- c("chr15", 56828384, 71672845);
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range, total.genes=11,
area.length=0.06, fixed.width=FALSE, gene.width=NULL);
track.pos <- RCircos.Get.Track.Positions(side="out", track.num=3);
track.height <- track.pos[1] - track.pos[2];
in.pos <- track.pos[2];
out.pos <- in.pos + track.height * 3;
RCircos.Plot.Zoomed.Ideogram.Ticks(zoom.info=zoom.range,
zoom.pos=zoom.pos, tick.interval=2, track.num=NULL,
inside.pos=in.pos, outside.pos=out.pos);
RCircos.Mark.Zoom.Area(zoom.range, track.num=NULL, zoom.pos=zoom.pos,
fill.color="green", inside.pos=in.pos, outside.pos=out.pos);
rm(zoom.range); rm(zoom.pos);
rm(track.pos); rm(track.height); rm(in.pos); rm(out.pos);
# Zoomed-in polygon plot with defined plot positions
data(RCircos.Polygon.Data);
data.col <- 4;
rows <- seq(1, 77, by=2)
plot.colors <- rep("red", nrow(RCircos.Polygon.Data))
plot.colors[rows] <- "green"
RCircos.Polygon.Data["PlotColor"] <- plot.colors
RCircos.Polygon.Data$Data[rows] <- RCircos.Polygon.Data$Data[rows]*-1
RCircos.Polygon.Plot(RCircos.Polygon.Data, track.num=5,
data.col=data.col, side="in")
data.rows <- which(RCircos.Polygon.Data$Chromosome=="chr9");
zoom.data <- RCircos.Polygon.Data[data.rows, ];
zoom.range <- RCircos.Get.Zoom.Range(zoom.data, genomic.columns=3);
zoom.pos <- RCircos.Set.Zoom.Plot.Positions(zoom.range,
total.genes=length(data.rows), area.length=0.08,
fixed.width=FALSE, gene.width=NULL);
track.pos <- RCircos.Get.Track.Positions(side="out", track.num=3)
track.height <- track.pos[1] - track.pos[2];
in.pos <- track.pos[2];
out.pos <- in.pos + track.height * 3;
min.value <- min(RCircos.Polygon.Data$Data)
max.value <- max(RCircos.Polygon.Data$Data)
RCircos.Plot.Zoomed.Polygons(zoom.data, data.col=data.col,
track.num=NULL, zoom.pos=zoom.pos, genomic.cols=3,
min.value=min.value, max.value=max.value, border.col=NULL,
inside.pos=in.pos, outside.pos=out.pos, outline=TRUE);
RCircos.Mark.Zoom.Area(zoom.range, track.num=NULL, zoom.pos=zoom.pos,
fill.color="green", inside.pos=in.pos, outside.pos=out.pos);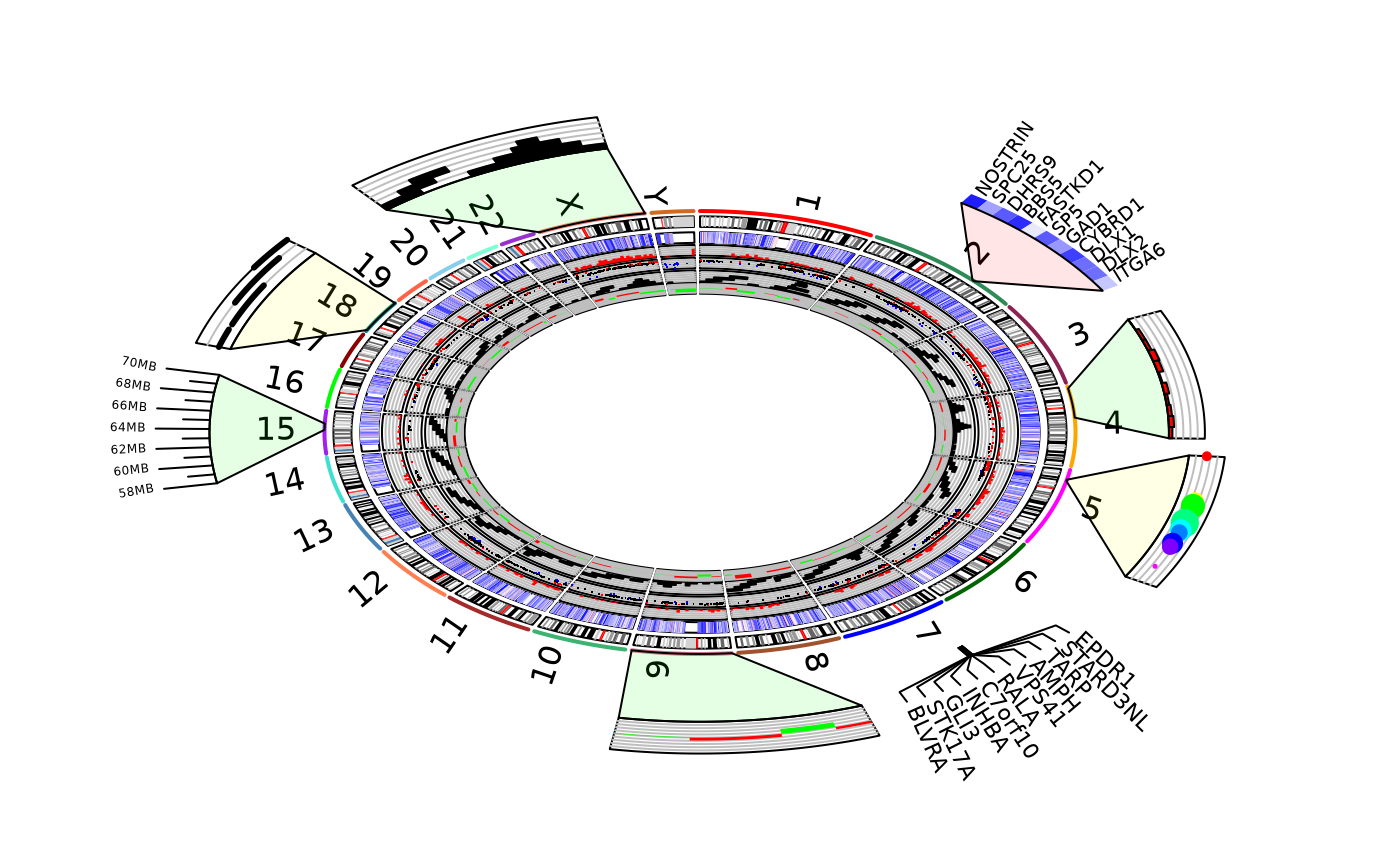
message("Zoom in plot demo done!")## Zoom in plot demo done!
# End of RCircos.Zoom.Plot.Demo.R